|
复旦大学附属儿科医院肝病科王建设教授带领博士后张晶和硕士研究生杨烨等,与复旦大学附属金山医院、上海医学院电镜中心、奥地利格拉茨大学医学院病理系合作完成的最新研究成果“Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: clinical, histological, and ultrastructural characterization” 近日于国际知名肝病杂志Liver International 在线发表。
低GGT胆汁淤积一般认为由基因突变引起。在1998年鉴定的ATP8B1缺陷引起的家族性胆汁淤积症(PFIC)1型和ABCB11突变引起的PFIC 2型约占低GGT胆汁淤积症的三分之二,三分之一病因未明。随着二代测序技术的进步,近些年相继又有TJP2缺陷病、FXR缺陷病相继被鉴定为低GGT胆汁淤积症的遗传学病因。2017年王建设教授团队鉴定了MYO5B缺陷引起的低GGT胆汁淤积症谱系,约占不明原因低GGT胆汁淤积症的20%,被国际上公认为家族性胆汁淤积症6型。然而仍有一部分低GGT胆汁淤积症病因未明,因此他的课题组继续致力于鉴定低GGT胆汁淤积症的新的“作案份子”。
为发现致病“新元凶”,该团队应用目前最先进的二代基因测序技术,先一次性高效筛查2703个基因(含64种已知遗传性胆汁淤积症致病基因)外显子区突变,并把通过该方法筛查后,仍不能明确病因的遗传性胆汁淤积症病例作为研究对象,用于 “新元凶”的鉴定工作。经过初筛,研究团队从16年来临床实践中积累的百余例遗传性低GGT胆汁淤积症病例中,筛选出44例遗传学病因不明的家系患儿,再进行“全外显子组”测序,一次性完成人类基因组所有近3万个基因编码蛋白区域(包括未知致病基因)的测序。经基因突变与致病可能性、临床症状契合度分析,研究团队最终在5个非近亲结婚家系的5例患儿中,发现“USP53基因”存在纯合或复合杂合“双突变”现象,即5例患儿的父母本身没有胆汁淤积症,但各携带了1个基因突变并遗传给了患儿,符合遗传学中隐性遗传病的遗传模式;患儿的其他健康兄弟姐妹则只携带1个基因突变,或没有携带该基因的突变,故没有发病。统计学分析表明,USP53基因的双突变与低GGT胆汁淤积症存在显着的相关性,占这批不明原因的低GGT遗传性胆汁淤积症患者的11%左右。随后临床工作中又陆续确认了2例USP53基因突变引起的婴儿低GGT胆汁淤积症患儿。至此,本次研究一共确认7例USP53基因双突变患儿,黄疸发作时间在生后2天~7月龄前。和以往鉴定的病例相比,七例患儿临床症状相对较轻,转氨酶活性、黄疸、瘙痒症状可以通过服用熊去氧胆酸和消胆胺等药物得到有效缓解和控制,目前尚无一例患儿需要肝移植。作者同时发现,USP53基因缺陷在不同患儿身上会对听力系统造成不同影响。患儿出生早期可伴有听力障碍,轻者可逐渐恢复,严重者会双耳失聪。本工作发现失聪最早可发生在1岁前,提醒要关注USP53基因缺陷患儿早期听力筛查及定期听力检测。
在本文撰写过程中,国外发表了一个家系,该家系为阿拉伯人,近亲结婚,三个患儿(2女1男)均为USP53基因纯合突变(该突变未在我们的患儿中发现)。三个患儿在4月龄~1岁间出现肝酶活性异常,低GGT胆汁淤积。服用利福平治疗对妹妹和堂弟肝酶活性恢复到正常水平、黄疸瘙痒消退有很好的效果,但对姐姐无效,姐姐6岁时因为难治性瘙痒接受了肝移植治疗。听力方面,3岁的堂弟没有耳聋症状,姐姐和妹妹分别在14岁和9岁出现了双耳失聪。鉴于有文献体外实验证明USP53和TJP2共定位,且USP53突变小鼠可导致耳聋,而人类TJP2缺陷可表现耳聋和/或低GGT胆汁淤积,由此提出USP53基因缺陷可能和耳聋及胆汁淤积相关。遗憾的是,该报道只有一个家系,且未提供USP53突变患儿的肝脏组织学证据。
王建设科研团队通过对USP53基因缺陷病患儿肝组织穿刺标本进行了详细观察和研究,进一步揭示了该病早期(10月龄)的组织学特征。光学显微镜下发现患儿肝组织内出现小叶紊乱、巨细胞、胆汁淤积、肝纤维化、肝硬化(图1),但未发现肝癌或胆管癌等癌变特征;免疫组织化学检测发现患儿肝组织中其他紧密连接蛋白复合体组分TJP2和CLDN1 有定位异常,蛋白表达量减少(图2);电镜下观察到肝细胞间的紧密连接结构变深变长(图3)。

图1. USP53 患儿 10月龄时肝脏病理特征
四位USP53患儿(P3,P4,P6和P7)均发现肝脏小叶紊乱和胆汁淤积。巨细胞(P3),肝纤维化(P3,P4,P7),肝硬化(P6)也在不同患儿中观察到。TJP2病人的病理图作为对照。
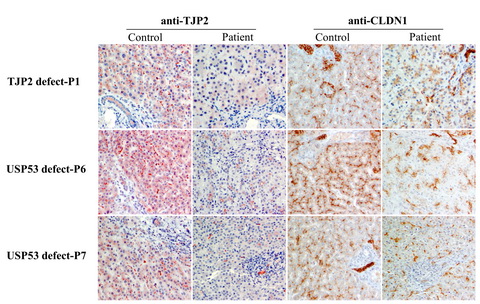
图2. USP53 患儿肝脏紧密连接组分蛋白TJP2和CLDN1的变化情况。
USP53缺陷病人中紧密连接蛋白组分TJP2和CLDN1 均出现表达量减少,定位异常,变化情况与TJP2缺陷病人类似。
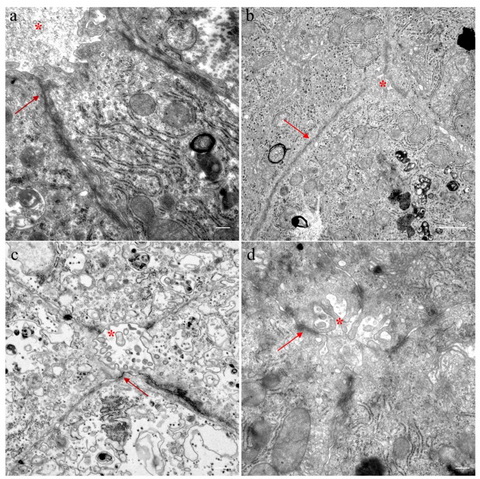
图3. USP53 患儿肝组织紧密连接结构变化。
与非紧密连接结构缺陷病人(ABCB11和ATP8B1,c 和d)相比,USP53缺陷病人的紧密连接结构变深变长。
以上工作明确了USP53基因双突变和胆汁淤积症的关联,且USP53基因缺陷病临床和组织学以及超微结构特征与TJP2基因缺陷病类似,提示都可能通过影响细胞间的紧密连接结构而引起肝脏问题和听力问题。由于有TJP2缺陷造成儿童期肝癌的报道,因此也提示我们要关注USP53基因缺陷患儿肝组织癌变倾向。
本工作确认了USP53突变导致的低GGT胆汁淤积症临床表型多样性,提示要密切关注患儿听力发育;首次提供了USP53遗传病婴幼儿期肝组织病变特征,提醒要关注和警惕肝组织是否有癌变;鉴于USP53 缺陷病与TJP2的临床表现高度相似,因此提示临床确诊需结合基因检测,从而使不明原因低GGT胆汁淤积症的诊断和治疗向个体化、精准化、精细化又迈进了一步。
本工作由国家自然科学基金81570468,81873543赞助。
本文链接:https://doi.org/10.1111/liv.14422 (doi: 10.1111/liv.14422)
参考文献:
[1] Qiu YL, Gong JY, Feng JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low gamma-glutamyltransferase cholestasis. Hepatology. 2017;65(5):1655-1669.
[2] Maddirevula S, Alhebbi H, Alqahtani A, et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet Med. 2019;21(5):1164-1172.
[3] Kazmierczak M, Harris SL, Kazmierczak P, et al. Progressive hearing loss in mice carrying a mutation in Usp53. J Neurosci. 2015;35(47):15582-15598.
[4] Walsh T, Pierce SB, Lenz DR, et al. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet. 2010;87(1):101-109.
[5] Wang HY, Zhao YL, Liu Q, et al. Identification of two disease-causing genes TJP2 and GJB2 in a Chinese family with unconditional autosomal dominant nonsyndromic hereditary hearing impairment. Chin Med J (Engl). 2015;128(24):3345-3351.
[6] Zou S, Mei X, Yang W, Zhu R, Yang T, Hu H. Whole-exome sequencing identifies rare pathogenic and candidate variants in sporadic Chinese Han deaf patients. Clin Genet. 2019;1-5.
[7] Sambrotta M, Strautnieks S, Papouli E, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46(4):326-328.
[8] Zhou S, Hertel PM, Finegold MJ, et al. Hepatocellular carcinoma associated with tight-junction protein 2 deficiency. Hepatology. 2015;62(6):1914-1916.
[9] Vij M, Shanmugam NP, Reddy MS, Sankaranarayanan S, Rela M. Paediatric hepatocellular carcinoma in tight junction protein 2 (TJP2) deficiency. Virchows Arch. 2017;471(5):679-683.
|

