|
近日,复旦大学附属儿科医院王艺教授领衔的SMA(脊髓性肌萎缩症)基因治疗研究团队,顺利完成II期临床研究的首批2例患儿入组给药,患儿经用药后7天安全性随访后已顺利出院。随访结果显示:患儿四肢肌张力较用药前明显提升,CHOP-INTEND评估分数增长明显,疗效开始显现;未发生2级及以上的药物相关不良事件,安全性良好。这其中一名越南籍患儿成为入组治疗的首例外籍患者,这也是我国基因治疗新药临床试验的首位国际小病友。

我国至今已上市的脊髓性肌萎缩症(SMA)靶向治疗药物仅两种,基因替代治疗药物Zolgensma尚未在中国上市且价格昂贵。
EXG001-307是由我国自主研发的新一代治疗SMA基因替代治疗药物,EXG001-307注射液作用机制和用法均与Zolgensma相似,有望一次给药长期有效。该临床研究也是国内相关领域首次尝试,研究由杭州嘉因生物科技有限公司申办,王艺教授作为主要研究者联合复旦大学附属儿科医院的多学科专家团队,组建了临床试验经验丰富的儿科专家为受试者随访及评估提供专业指导。
这名越南籍SMA患儿的父母,通过美国家属获得研究信息后,抱着一线希望辗转联系上复旦儿科医院王艺教授研究团队。王艺教授和研究团队第一时间给予了家属积极反馈。研究团队面临一系列需要解决的问题:如何让外籍患儿顺利抵达研究中心入组临床研究、如何突破研究的国界限制并符合伦理要求等。最后在医院多方协助支持下,问题得到快速稳妥解决,患儿家属带着期许踏上前来中国的求医之路。
抵达上海时,患儿出生已5个月零20天,距离“用药当天年龄不超过出生后第180天”的入组条件已非常接近。“你跨越山海而来,我必倾心倾力以待”,王艺教授及团队迅速协调各方力量,加速院内流程,与时间赛跑,仅用1周时间即完成对该名外籍患儿的筛选、入组并顺利用药。
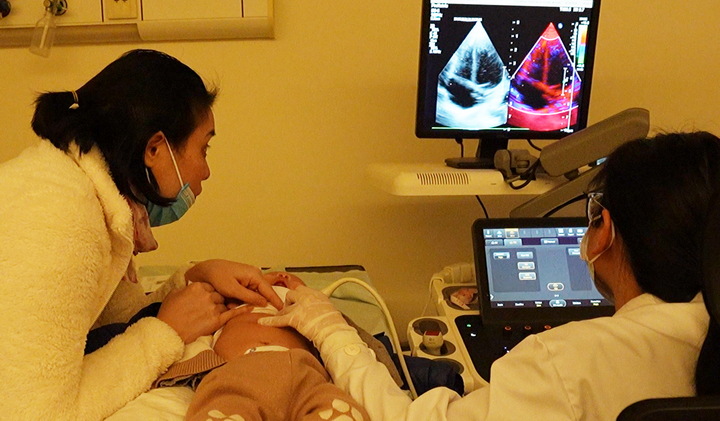
此例外籍患儿的入组,突破了临床研究国界限制,跨越语言鸿沟,打开了我国SMA基因治疗研究的国际大门,为我国罕见病临床研究开辟新的思路和方式,也加速了罕见病药物的临床阶段,对罕见病药物的快速上市造福更多患者具有重大意义。
据介绍,随着临床研究的持续顺利推进,王艺教授SMA研究团队已陆续在“中华医学会第28次全国儿科学术大会”以及美儿SMA关爱中心等官方平台汇报分享初步的试验进展。截至目前,所有接受治疗的1型SMA患儿,安全性、耐受性持续表现良好,无死亡事件,无剂量限制性毒性,无>2级与药物相关不良事件。接受治疗后的患儿身体各项运动功能提升明显,其中有一半患儿在用药后1个月的随访中即实现了“头部保持直立”;最早接受治疗的患儿已实现了无他人支撑独坐,独站、行走等指日可待;高剂量组的首例患者在不满6月龄时已可“扶坐”。
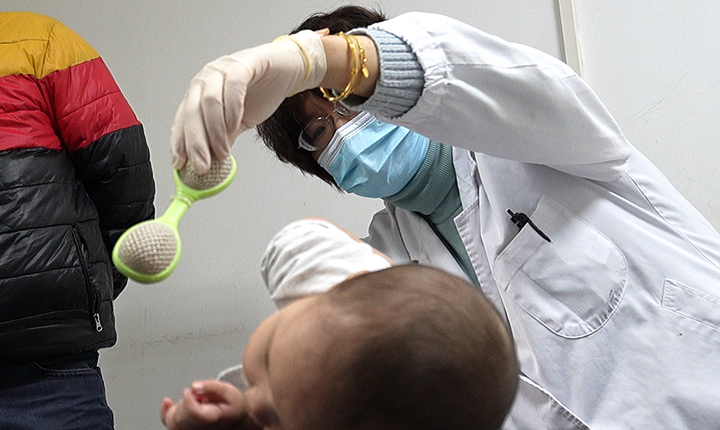
目前该研究已完成I期剂量“爬坡”,II期扩展临床研究仍在持续招募1型SMA患儿,有需要的患儿家庭可联系复旦儿科相关团队。
SMA基因治疗II期临床研究患者招募中
招募对象:1型脊髓性肌萎缩患者
尊敬的患者家属:
目前复旦大学附属儿科医院正在进行一项治疗1型脊髓性肌萎缩的临床研究。该研究已获国家药品监督管理局批准,临床试验批准通知书编号为:2022LP00989。
目前正在为这项“一项在1型脊髓性肌萎缩患者中评估静脉注射EXG001-307后的安全性和有效性的多中心、单臂、非随机、开放性临床试验(方案编号:EXG001-307-102)”招募患者;该研究预计招募9-24例1型脊髓性肌萎缩患者;该研究已获得复旦大学附属儿科医院伦理委员会批准,同意进行此项临床研究。
主要入选条件:
1)年龄不超过出生后第180天;
2)经双侧等位基因SMN1突变(缺失或点突变)基因诊断为SMA;具有2个拷贝SMN2基因;
3)临床病史、体征符合I型SMA表现,即肌张力减退,运动功能发育滞后、头部控制不佳、圆肩姿势和关节过度活动;
主要排除条件:
1)出生时胎龄小于35周(245天)
2)筛选期时,在清醒或睡眠且未接受任何辅助供氧或呼吸支持时,血氧饱和度<96%。
3)需要有创通气或气管切开术,或当前使用无创通气支持平均≥16小时/天。
4)筛选前2周内患有严重非呼吸道疾病;
5)既往使用过其他SMA治疗药物(如诺西那生纳、利司扑兰及Zolgensma等)或参加过其他SMA治疗药物的临床研究(包括但不限于上述3种药物)。
如果您想详细咨询或参加本项目,请随时与研究医生联系:
复旦大学附属儿科医院联系人:朱医生
联系电话:18101843510
地址:上海市万源路399号
|


