|
复旦大学附属儿科医院感染传染科王建设教授团队最新研究成果《ABCB11缺陷引起暂时性新生儿肝内胆汁淤积症及其与基因型和肝脏BSEP表达的相关性》“ABCB11 deficiency presenting as transient neonatal cholestasis: Correlation with genotypes and BSEP expression” 近日于国际知名肝病杂志Liver International (《国际肝病》)(IF=5.175)在线发表。这是该课题组ABCB11缺陷病相关成果第3次刊登在该杂志。

ABCB11基因编码胆盐外运泵(bile salts export pump, BSEP)蛋白。1998年该基因被鉴定为进行性家族性肝内胆汁淤积症2型(PFIC2)的致病基因,临床表现为低GGT胆汁淤积症。目前国际上公认ABCB11双等位基因突变有2种临床表型:PFIC2和良性复发性肝内胆汁淤积症2型(BRIC2)。PFIC一般在婴儿期发病,胆汁淤积症持续性/进行性加重,常导致儿童期死亡或需要肝移植才能长期存活;BRIC发病较晚,多在几岁至十几岁,甚至成年发病,胆汁淤积发作期与缓解期交替,发作间期肝脏完全正常。新论文确立了ABCB11双等位基因突变引起的第三种临床表型:暂时性新生儿胆汁淤积症(TNC),胆汁淤积消退后不再复发,肝功能持续正常。
王建设教授从2003年开始进行ABCB11缺陷病的研究,在医院和国家自然科学基金面上项目的支持下,先后对24例临床上表现为低GGT婴儿进行性胆汁淤积症病例进行ABCB11基因测序,最终7例基因确诊ABCB11双等位基因突变,这一发现由刘丽艳硕士为第一作者于2010年发表于Liver International (Liu LY, Wang ZL, Wang XH, et al. ABCB11 gene mutations in Chinese children with progressive intrahepatic cholestasis and low glutamyltransferase. Liver Int. 2010,30(6):809-815)。这是课题组第一次在该期刊发表论文,也是国际上首次报告华人中该病的基因突变特征,因此该刊同期配发了对该文的评论。
随后的10余年间,研究团队继续累积病例,并追踪随访。在国家自然科学基金国际(地区)合作与交流项目-中国加拿大健康合作计划支持下,刘腾博士采用高效液相色谱质谱方法对ABCB11缺陷病患儿的血清胆汁酸代谢谱进行分析,发现了多种胆汁酸代谢相关的代偿机制,提示胆汁酸代谢谱与患儿表型密切相关,并可以反映胆汁酸在体内的合成代谢及肠肝循环状态。该研究成果于2018年发表于Liver International (Liu T, Wang RX, Han J, et al. Comprehensive bile acid profiling in hereditary intrahepatic cholestasis: Genetic and clinical correlations. Liver Int 2018;38:1676-85. )。
最新发表的论文收集了感染传染科王建设教授团队累计确诊的68例ABCB11双等位基因突变患儿,汇总了ABCB11基因突变谱、生化指标及肝组织BSEP免疫组化特征和长期随访的预后资料。68例患儿中,42例表现为PFIC,3例表现为BRIC,23例黄疸消退后在随访期内胆汁淤积症未复发,符合TNC的诊断。以往临床实践中通常看到婴儿期起病的ABCB11双突变就会诊断为PFIC,本文确立了ABCB11双等位基因突变的又一新表型,因此对基因诊断报告的解读有重要意义。
那么哪些患儿会表现为TNC呢?通过和PFIC患儿比较发现,两组患儿发病年龄和首诊时肝功能化验指标无明显差异,因此不能预测ABCB11双等位基因突变患儿的预后。作者进一步通过基因型-表型分析,发现TNC患儿携带双等位基因non-null(预测不会导致蛋白完全无表达)变异的比例(17/23)显著高于PFIC (20/42, P = 0.041)。TNC患儿肝脏表达BSEP蛋白的比例(7/7)也显著高于PFIC(5/16, P = 0.005)。同时Kaplan-Meier分析显示肝组织有BSEP表达的患儿自体肝生存时间更长,预后较好(P = 0.009)。由此得出结论,在ABCB11双等位基因突变引起的新生儿胆汁淤积症中,ABCB11基因型和肝组织BSEP表达情况决定了相关婴儿胆汁淤积症的临床表型是PFIC,还是TNC表型,对于个性化预测ABCB11双突变患儿的预后有指导意义。
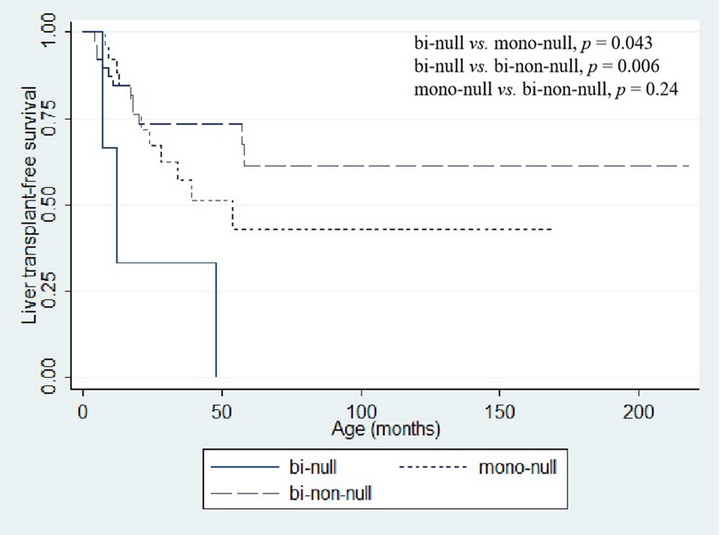
Fig. 1 Kaplan-Meier plot, liver transplant (LT)-free survival: Genetics (N bi-null = 3, N mono-null = 26, N bi-non-null = 39)
ABCB11 variants and survival without LT. 100% (3/3) of patients with biallelic null variants in ABCB11, 46% (12/26) with a monoallelic null variant paired with a monoallelic non-null variant, and 31% (12/39) with biallelic non-null variants underwent LT or died of liver failure.

Fig. 2 Kaplan-Meier plot, liver transplant (LT)-free survival: Expression of bile salt export pump (BSEP)
(NBSEP absent = 12, NBSEP expressed = 12, P = 0.009)
BSEP expression and survival without LT. 67% (8/12) of patients without BSEP expression and 8% (1/12) patients with BSEP expression underwent LT or died of liver failure.
王建设教授团队从诊断国内第一例ABCB11双等位基因突变导致的家族性进行性肝内胆汁淤积症到ABCB11双等位基因突变暂时性胆汁淤积症新表型文章的发表正好经历了18个年头。18年中,要感谢医院和多项国家自然科学基金项目的支持,感谢科室前辈打下的良好基础,感谢科室同事和众多合作者的鼎力相助,让复旦大学附属儿科医院在遗传性胆汁淤积性肝病诊断方面的目标从追赶国际先进水平逐渐到达国际领先水平。
最新发表的论文第一作者是李丽婷博士后,她于2011年加入课题组,追踪随访ABCB11缺陷病患儿近10年,付出了艰辛的努力。也要感谢一路相伴走过的患者及家属,他们见证了复旦大学附属儿科医院儿童肝病课题组的成长,从对该病的一无所知进步到能对该病做出基因和功能方面的精准诊断。
王建设教授是3篇论文的通信作者。他表示课题组的下一个目标是ABCB11缺陷病的精准治疗。首先对患者根据课题组在基因型-表型关系、病理免疫组化和表型关系、胆汁酸谱和表型关系方面研究成果的基础上对ABCB11双等位基因突变引起的家族性胆汁淤积症患儿进行分型;然后根据不同突变的致病机制不同引入不同的分子靶向治疗治疗,希望为ABCB11缺陷病儿童创造更好的明天。
|


